

While Post Market Surveillance (PMS) is mentioned in Annex X of the existing Medical Device Directive (MDD), it was not defined clearly in the MDD. Lack of clarity is completely removed with the publication of the new EU MDR. Not only is PMS defined in Article 2 (60), it is listed as one of the general obligations of all manufacturers (new Article 10). It is also one of the topics specifically called out for monitoring by the person responsible for regulatory compliance (new Article 15). Post Market Surveillance, as described in EU MDR Medical Device Regulation, is a new concept. A manufacturer shall know at all times how his product performs in the field.For each device, manufacturer shall establish a post market surveillance system (PMS system).The PMS system has to allow:
- Systematic and active gathering of information;
- Cooperation with the competent authorities responsible for vigilance and market surveillance;
- Connection with the system for corrective action or preventive action to incorporate lessons learned; and,
- Update of the technical documentation, including the risk-benefit determination and clinical evaluation
This system shall be an integral part of the manufacturer’s QMS and shall be based on a post market surveillance plan (PMS plan). The requirements of the post market surveillance plan are set out in Annex III. 1.1. That PMS plan shall be part of the technical documentation. Mainly, the PMS system shall be a proactive process and shall be constantly updated. (Article 83 & 84).
Post-market surveillance plan requirements
Under the Regulations, a PMS plan has to be established for each device or device family. Potential information for use in PMS comes from a number of sources, including:
- investigations of serious incidents, investigations of incidents not meeting the criteria for classification as serious incidents, data on undesirable side effects;
- trend analysis and reporting;
- field safety corrective actions;
- reports in specialist or technical literature;
- reports or outputs from databases or registries;
- complaints provided by users, distributors and importers;
- other feedback including customer surveys, information provided as input into manufacturer’s websites and reports in social media; and,
- publicly available data on events with similar devices provided by other manufacturers.
PMS plan includes a description of indicators and thresholds for continuous reassessment of risk management and the risk-benefit analysis together with the means to:
- investigate complaints and market experience from the field;
- monitor trends, identify statistically significant increases in frequency or severity of incidents and provide trend reports;
- communicate with competent authorities and notified bodies;
- communicate with authorized representatives, importers, distributors, users and patients;
- trace and identify devices for which correction or corrective action might be necessary;
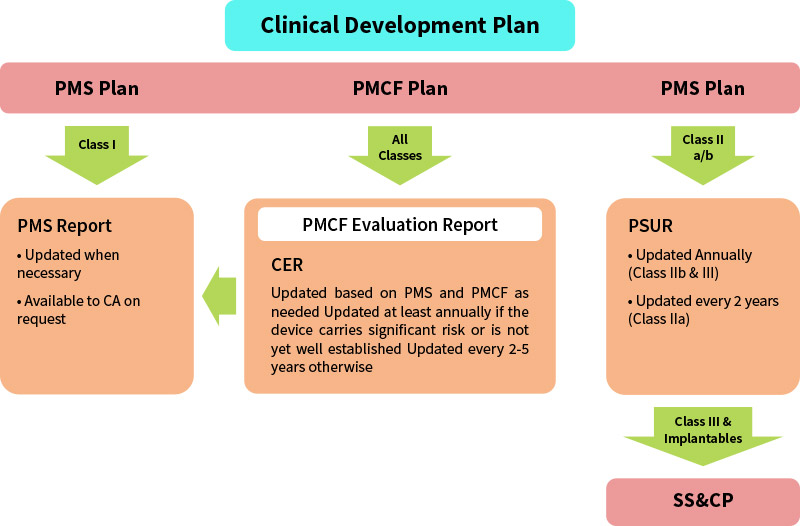
The PMS plan should also reference the documented procedures describing i) the PMS system, ii) the creation of the PMS and PMCF Plan, iii) the generation of the Periodic Safety Update Report (PSUR) or PMS report, as applicable; and, iv) the processes for identification and implementation of corrections, corrective actions or preventive actions. The PMS plan is an element of the technical documentation and so needs to be in place before the Declaration of Conformity to the applicable Regulation is drawn up and before the device can be CE marked under the Regulations.
| Proactive | Reactive |
|---|---|
| Customer surveys | Customer complaints |
| Post CE mark clinical trials , including PMCF | Unsolicited user feedback (other than complaints) |
| Manufacturer sponsored device tracking/implant registries | Maintenance/service reports |
| Expert user groups (focus groups) | Trend analysis |
| Market Actions (FSNs) | Failure analysis |
| Adverse event reports | Social media |
| Literature reviews |
The Regulations contain new requirements to prepare summary reports of PMS information for all classes of devices.
| Class I | Class IIa | Class IIb | Class III | Implantable | |
|---|---|---|---|---|---|
| PMSR | PSUR | throughout the lifetime of the device | PMCF | ||||
| As necessary | As necessary or every 2 years | As necessary or annually at a minimum | |||
| Available to the Notified Body (NB) | Submitted to NB via EUDAMED | ||||
| Available to Competent Authority upon request of Notified Body | Made available to Competent Authority via Electronic filing EUDAMED | ||||
Some Challenges
- As PMCF plan to be part of PMS plan, currently organizations do these independently and PMS (or Quaity teams) are siloed from Clinical teams in some companies. As PMS team needs to sign off the PMS plan, dependency or alignment with Clinical is important going forward
- One of the newer requirements is Literature Search. Most PMS teams neither have processes nor teams to do this activity. Teams need to plan on what and how to do the Literature part
- The design of a quality management system which is conducive to the potential of multiple updates to processes, technical documents and even devices themselves
- Establishment of suitable indicators and threshold values that shall be used in the continues reassessmentof the benefit risk analysis and of the risk management
- Establishment of effective and appropriate procedures describing PMS system, PMS plan, PMCF plan, CAPA, trend investigation
Achieve a comprehensive understanding of PMS Planning and Challenges under EU MDR by partnering with industry experts. Keep yourself updated. Ensure compliance.
