

In March 2024, The document “Regulatory Guidance for Software Medical Devices – A Lifecycle Approach” whose third revision was issued by the Singapore Health Sciences Authority. The document is intended to cover software of all Risk Classifications, addressing regulatory requirements throughout the entire product life cycle. It specifically focuses on key software-related regulatory aspects including cybersecurity and requirements for Artificial Intelligence (AI) medical devices.
The document will address the following topics:
- Quality Management System (QMS) for software medical devices
- Pre-market product registration requirements
- Dealer’s licensing requirements
- Change notification
- Post-market management of software medical devices
- Cybersecurity
- Artificial Intelligence
Quality Management System (QMS) For Software Medical Devices
All manufacturers of medical devices, including software medical devices, are required to implement a Quality Management System (QMS) to ensure consistent manufacturing quality. For software medical devices, good software quality and engineering practices is essential to control the quality of software products. The international standard ISO 13485 – Medical Devices – Quality Management Systems – Requirements for regulatory purposes outlines the requirements for a QMS that can be adopted by organizations involved in any stage of the medical device life cycle.
Pre-Market Product Registration Requirements
Product registration applications for medical devices submitted to HSA must conform to the format specified in the ASEAN Common Submission Dossier Template (CSDT) document. This format can be adapted from the International Medical Device Regulators Forum (IMDRF) Non-in Vitro Diagnostic Medical Device Market Authorization Table of Contents (nIVD MA ToC). The mapping between the pertinent sections in the IMDRF ToC dossier and CSDT is available at https://www.hsa.gov.sg/medical-devices/guidance-documents.
This section provides guidance for specific sections of the CSDT dossier where there may be particular requirements for software medical devices. Following are the sections addressed here:
- Essential Principles for safety and performance of medical devices
- Labelling requirements
- Software versioning and traceability
- Software verification and validation
- Clinical evidence
- Risk management
- Supporting documents for cybersecurity
Software Manufacturers and Distributors: Activity Controls
All manufacturers, importers, and/or wholesalers of software medical devices must hold medical device licenses for their respective activities. A prerequisite for obtaining a license is the establishment and maintenance of an appropriate Quality Management System (QMS), which should cover the following aspects:
- Ensure that the software is developed and manufactured within an appropriate and effective quality management system (e.g., ISO 13485).
- Ensure traceability of the software medical device, essential for tracking and tracing the software (e.g., software version) to users (e.g., physicians or patients) in the event of a Field Safety Corrective Action (FSCA) or product defect.
- Provide assurance that proper procedures are in place for post-market surveillance and response, including the ability to handle product recalls and implement corrective actions (e.g., bug fixes, cyber alerts, software patches) in a timely and effective manner (planning, conducting, and reporting of corrective action), and to identify any recurring problems requiring attention.
- Ensure proper maintenance and handling of device-related records and information (e.g., customer complaints, distribution records, recall data) throughout the lifecycle of the software.
Do note that for all the scenarios mentioned in the document, the software medical device will require product registration.
Changes to A Registered Software: Change Notification
A software medical device undergoes a number of changes throughout its product life cycle. These changes are typically intended to (i) correct faults, (ii) improve the software functionality and performance to meet customer demands and (iii) ensure safety and effectiveness of the device is not compromised (e.g. security patch).
For instance, significant changes, such as Technical & Review changes, will necessitate a more comprehensive review compared to Administrative changes to ensure that the alteration does not compromise the safety and effectiveness of the software. As such, non-significant software changes are required to be notified to the HSA and are referred to as Notification changes.
Below are some common examples of changes that would require approval from the HSA before implementation.
A Technical Notification will be required for Class C and D products, and a Notification will be required for Class B products if:
- Alters an algorithm
- Addition of new features or software applications
- Addition or removal of alarm function
- Change in the operating system
All other changes, including minor adjustments to rectify errors or address cybersecurity vulnerabilities, would require a notification.
Post-Market Management of Software Medical Devices
This section provides an overview of some post-market requirements that are also applicable to software medical devices.
Field Safety Corrective Actions (FSCA)
An FSCA may be initiated when the product owner identifies specific risks associated with the use of the medical device during post-market monitoring and surveillance, such as through tracking product complaints or feedback. The purpose of initiating an FSCA is typically to communicate these risks to users and to outline the measures planned to mitigate them.
Adverse Events
Reports may come from various sources including surveillance of device log sheets, complaints or feedback from the user. It is crucial to promptly investigate these reports and implement corrective and/or preventive actions in a timely manner to effectively manage risks and prevent the recurrence of adverse events.
Software with Multiple Functions
Software medical devices typically consist of various functions, some of which may not meet the criteria outlined for a medical device in the Health Products Act (HPA). These non-medical device functions may include the following:
- Software function that allows storing, converting formats or transferring patient data;
- Software function that is intended to provide general patient education and facilitate access to commonly reference information;
- Software function that allows automation of general office operations (e.g. patient scheduling, billing and etc.) in a healthcare setting.
Cyber Security
Cyber Security Considerations
When developing a software medical device, it’s essential to devise a cybersecurity plan that includes the following considerations (non-exhaustive):
- A secure device design
- Having proper customer security documentation
- Conduct cyber risk management
- Conduct verification and validation testing and
- Having an on-going plan for surveillance and timely detection of emerging threats
Artificial Intelligence Medical Devices (AI-MD)
This section addresses specific regulatory considerations pertaining to medical devices integrating Artificial Intelligence (AI) technology from a medical device regulatory perspective.
Regulatory Requirements for AI-MD
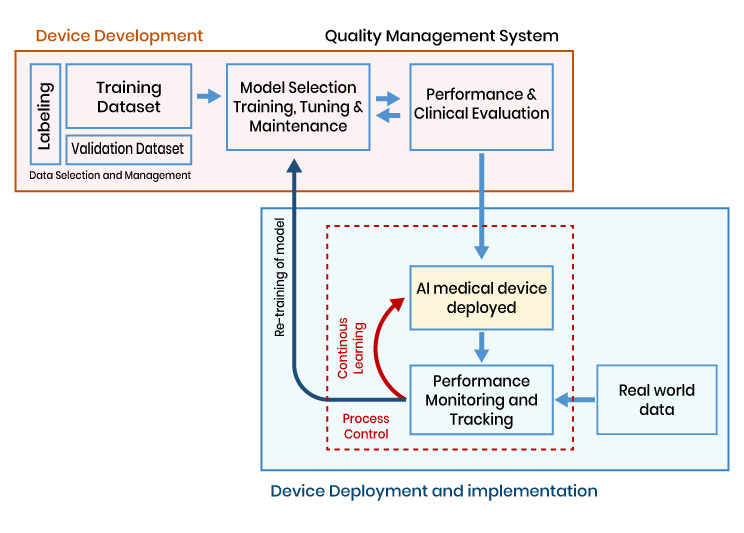
The regulatory principles for AI-MDs are comparable to software that are regulated as medical devices However, there are distinct additional considerations, such as continuous learning capabilities, the level of human intervention, model training, retraining, etc., which demand meticulous attention and tailored approaches when addressing the regulatory requirements for AI-MDs.
The block diagram below illustrates the process of developing and deploying the AI-MD (Artificial Intelligence in Medical Diagnosis).
MakroCare offers extensive Regulatory assistance for manufacturers looking to enter the Singapore markets with their medical devices. Need full Regulatory support? Contact us to schedule a call today!
