

Statistical monitoring (SM) involves the review of ongoing prospective study data and it involves scanning for odd or clustered data patterns that may be indicating problems with the study’s data integrity, quality, or safety of the participants.
The International Council for Harmonization (ICH) approved the updated E6 guideline, identified as “Integrated Addendum to Good Clinical Practice (GCP),” on December 15, 2016. The same national/regional procedures that apply to other regulatory requirements and guidelines are now used for regulatory implementation (ICH 2017).
Importance of Statistical Monitoring in ICH E6 (R2) Addendum
- Incorporating a risk-based approach into clinical trials.
- It helps to guide decisions surrounding investigations into data discrepancies and identifying where site directed monitoring visits may be necessary.
- Help companies to ensure the safety and quality of clinical trials by identifying and mitigating risks, as well as ensuring that any incorrect data is identified, in the early stages.
- Risk-based monitoring is becoming more popular as it is recognised as a cost effective and efficient method of data analysis.
- Centralizing statistical monitoring, as opposed to on-site source data verification, can make risk-based monitoring easier as it reduces the need to visit all clinical trial sites for data analysis and verification as it can be outsourced to one supplier, reducing time and resource.
- Statistics play a key role in analyzing and making decisions on study support pharmaceutical and biotechnology companies as they opt to centrally monitor their data and introduce risk-based approaches into their study designs.
Monitoring
The purpose of monitoring is to protection of rights and wellbeing of human participants ensure consent in place for record access. Trial data must be accurate, complete and verifiable from source documents. Conduct of trial is in compliance with currently approved protocol, GCP and all applicable regulatory requirements.
Monitoring of clinical trials is a fundamental process required by regulatory agencies. It assures the compliance of a center to the required regulations and the trial protocol. Multicenter clinical trials are imperative to obtain a conclusive assessment concerning the safety and efficacy of medical treatments. They involve diverse clinics or hospitals, and their respective personnel. This requires the monitoring team to ensure the compliance of each center to the study protocol and the requirements of good clinical practice.
Types of Monitoring
Central Monitoring
Determination of key eligibility criteria through collection of Consent forms, scans, pathology reports. In statistics unusual patterned of data.
Centralized monitoring processes provide additional monitoring capabilities that can complement and reduce the extent and/or frequency of on-site monitoring and help distinguish between reliable data and potentially unreliable data.
The concept of centralized monitoring is centered around three pillars:
- Data Surveillance
- Key Risk Indicators (KRIs)
- Quality Tolerance Limits (QTLs)
Centralized Statistical Surveillance
The new regulation asks both sponsors and CROs to raise perspectives on the whole study – from the beginning and throughout the trial – to conduct a more holistic valuation of risk and then adjust methods of oversight at each site accordingly.
The goal of Data Surveillance is to detect quality issue and potential study misconduct by identifying atypical patterns and anomalies in the data. The key tenet is that the sites participating in a given study have a common database structure inherited from the CRF design. This makes it possible to compare sites and identify those whose data are inconsistent with others. Methods relying on the use of large number of statistical tests are particularly effective in identifying unexpected or unusual patterns in trial data. Various forms of study misconduct can be detected with this approach, from intentional data fabrication or alteration, to site sloppiness and training issues.
It enables sponsors to identify issues such as a lack of variability or implausible values that may go undetected using other approaches. Detecting Key Risk Indicators (KRI) is major task for statisticians in coordinating with Clinical Research Team at site and Clinicians.
Based on actual clinical data: all patient-related variables and endpoints in a study are deemed to be indicative of quality.
A risk score can be assigned to each site with an advanced and sophisticated statistical approach.
Results may be presented both with a statistical report or by using an interactive software (such as SAS Visual Analytics) to summarize anomalies detection with plots.
An application of the analysis that can be implemented in Visual Analytics is presented next.
Centralized Statistical Surveillance is an unbiased approach, fully data driven, which means that any “red flags” are governed by statistical tools that have detected discrepancies in the data. No arbitrarily fixed thresholds are involved in contrast to other risk-based monitoring systems. Hence, central monitoring through advanced statistical techniques can detect abnormal/anomalous patterns in the data. It can also help improve the effectiveness of on-site monitoring by prioritizing site visits and by guiding site visits with central statistical data checks.
Risk Based Monitoring
In RBM limited resources should be used to where it is really necessary the main high risk part is for subject protection, reliability of trial results, protection of future patients and Risk based approach.
The increasing complexity of clinical and post-market trials has put pressure on all involved to focus on activities that will ensure the accuracy necessary to meet regulatory approvals. As the clinical phase is the most complex part in a drug development process, it requires keen attention on efficient planning, conducting and monitoring of trials to achieve reliable study data appropriate for submission. And, along with the complexities associated with data collection has come higher costs to the sponsor in both trial management and clinical monitoring to achieve that higher data quality and thus, better patient safety.
As a guidance for the industry, regulatory authorities have recommended that sponsors and CROs rely more heavily on centralized monitoring practices to promote risk mitigation and early issue detection to improve data quality and patient safety in a more cost-efficient manner.
This recommendation comes from studies that indicate the traditional method of on-site monitoring using source data verification (SDV) has been discredited in that it is not as effective as once perceived. In fact, according to statistics, SDV only touched about 2% of the data and only changed 1%. With the costs involved in manual verification and the substandard results, industry regulators are reassuring a new look at how data is collected and verified.
On Site Monitoring
Staff training, access to necessary documents, confirm pharmacy and lab resources in place, Adherence to protocol and GCP check medical records and Source Data verification.
Discussion
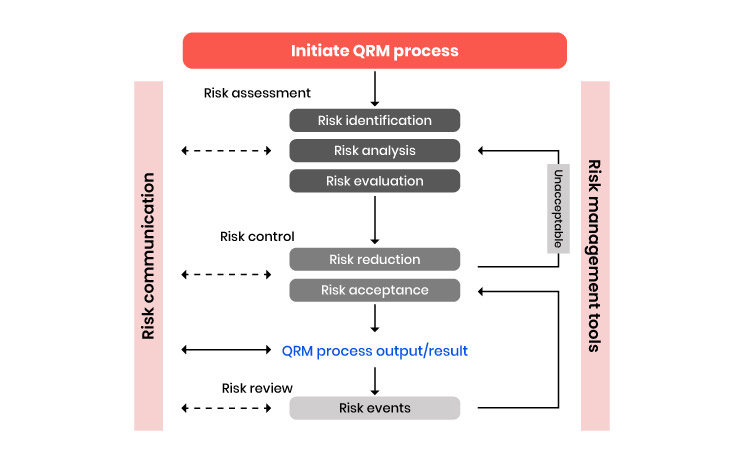
Effective monitoring is critical to ensure both subject protections and high quality trial data. Monitoring continues to be the sponsor’s responsibility. By far, the most substantial changes to ICH E6 are related to study monitoring. The addendum incorporates elements from the FDA’s (2013) risk-based monitoring guidance, which supports alternative approaches (specifically, risk-based and combination activities) to monitoring. The revised ICH E6 requires that the sponsor develop.
A systematic, prioritized, risk-based approach to monitoring clinical trials. The flexibility in the extent and nature of monitoring described in this section is intended to permit varied approaches that improve the effectiveness and efficiency of monitoring. The sponsor may choose on-site monitoring, a combination of on-site and centralized (off-site) monitoring, or, where justified, centralized monitoring (only). The sponsor should document the rationale for the chosen monitoring strategy (e.g., in the monitoring plan).
The ICH E6 addendum defines centralized monitoring and distinguishes it from on-site monitoring. Centralized monitoring allows the real-time review of accumulating trial data, which helps to identify missing or inconsistent data, examine trends, identify data errors, analyze sites/investigators, and/or select sites for targeted on-site monitoring.
Per section 5.18.6(e), “monitoring reports,” including both centralized reports and on-site monitoring visit reports, are now required to be provided to the sponsor (including appropriate sponsor management and CRO staff) by the monitor in a timely manner and with sufficient detail to allow sponsors to follow up, if needed. This allows and requires the sponsor to follow-up on identified serious noncompliance. In section 5.20, the addendum adds the sponsor should perform a root cause analysis and implement appropriate corrective and preventive actions (for example, a corrective and preventative action plan) if noncompliance is or may be serious. Finally, each study now requires a study-specific monitoring plan. The plan should take into consideration potential risks of harm to human subjects and data integrity. The monitoring plan should not only include how the study will be monitored, but a rationale. Additionally, the monitoring plan should also emphasize the monitoring of critical data and processes, especially those that are not routine clinical practice and require extra training (ICH 2016).
ADDENDUM as per the guidelines in monitoring report
The Reports of on-site and/or centralized monitoring should be provided to the sponsor (including appropriate management and staff responsible for trial and site oversight) in a timely manner for review and follow up. Results of monitoring activities should be documented in sufficient detail to allow verification of compliance with the monitoring plan. Reporting of centralized monitoring activities should be regular and may be independent from site visits.
Monitoring plan: The sponsor should develop a monitoring plan that is tailored to the specific human subject protection and data integrity risks of the trial. The plan should describe the monitoring strategy, the monitoring responsibilities of all the parties involved, the various monitoring methods to be used, and the rationale for their use. The plan should also emphasize the monitoring of critical data and processes. Particular attention should be given to those aspects that are not routine clinical practice and that require additional training. The monitoring plan should reference the applicable policies and procedures.
Conclusion:
As pharmaceutical and biotechnology businesses choose to centrally monitor their data and incorporate risk-based techniques into their research designs, statistics play a critical part in the analysis and decision-making process related to study data. ICH E6 R2 relies on centralized and unsupervised statistical assessments of data quality, giving sponsors and CROs alike the chance to concentrate efforts on what counts most. CROs and pharmaceutical corporations are not the only organizations that employ these kinds of procedures to analyze data using statistical methodologies. The FDA is aggressively looking at data analysis to identify places that are at risk for inspection, demonstrating interest from the regulatory community as well.
