

Benchmarking is a formal procedure of evaluation in a process or system preferably quantitative, but sometimes necessarily qualitative. Medical device industry plays a critical role in advancing healthcare globally, and it is crucial to guarantee patient safety and product efficacy. To guarantee that their products satisfy the highest standards, medical device manufacturers operating in the European Union (EU) are subject to a number of rules. The EU In Vitro Diagnostic Regulation (EU IVDR) and the EU Medical Device Regulation (EU MDR) are the two main regulations controlling medical devices in the EU.
EU Medical Device Regulation (EU MDR):
The EU MDR considerably reinforced the regulatory framework for medical devices in the EU and superseded the old Medical Device Directive (MDD), which went into effect on May 26, 2021. The EU MDR considerably broadens the scope of products covered by the regulation by introducing new definitions and classifications for medical devices in comparison to the MDD. This growth presents a challenge for manufacturers trying to prove compliance. To demonstrate compliance with all pertinent provisions of the Medical Device Regulation (EU) 2017/745, one must demonstrate EU MDR compliance. For manufacturers to receive regulatory approval for their medical devices, they will have to prove compliance.
According to EU MDR regulations, medical device manufacturers must create a set of compliant regulatory systems, procedures, and documentation to continuously assess the performance and safety of their products. Medical device manufacturers must also design, manufacture, distribute, and track their products in accordance with these criteria.
Complying with the EU MDR requires manufacturers to have a thorough awareness of the regulations and their obligations.
The EU MDR’s extensive reach Section 10.4.1:
The EU Medical Device Regulation (EU MDR), specifically Section 10.4.1, is essential for guaranteeing the safety and quality of medical devices. Manufacturers must list particular substances in their products according to this regulation. Manufacturers are specifically required to identify reproductive toxins (CMRs), mutagens, carcinogens, and endocrine disruptors chemicals (EDCs) that are present in invasive devices or materials that come into contact with fluids or gases. If these compounds are found in amounts higher than 0.1% w/w, they need to be identified.
Medical device manufacturers are subject to a number of important compliance obligations under the EU MDR. First and foremost, manufacturers must place their devices into one of four classes—Class I, Class IIa, Class IIb, or Class III—according to the severity of risk associated with them. The appropriate level of scrutiny and conformance evaluation processes are determined by this classification.
According to EU MDR Annex VIII, the classification process is based on 22 rules. Rules 19–22 are new to the EU MDR, while rules 1–18 have been carried over from the previous MDD. The impact of the EU MDR on products can be determined by companies taking into account all three criteria referred to in section 10.4.1, together with those classification rules.
A complete technical documentation, which contains detailed information on the design, manufacture and performance of a device, should also be drawn up and maintained by manufacturers. Compliance with the general safety and performance requirements set out in this Regulation should be demonstrated by these documents. In order to support the safety and performance claims of the device, it should also include a clinical evaluation report (CER) based on collected clinical data.
The requirement for notified bodies is another key element in EU MDR compliance. In order to assess the conformity of medical devices, notified bodies are independent organizations established by EU Member States. In order to demonstrate compliance with the Regulation, manufacturers must obtain a valid certificate from the notified body. The certification process is based on a thorough assessment of the technical documentation, quality management system and in some cases an examination of clinical evidence.
Main components of EU MDR Compliance
The EU MDR compliance can be assessed in three main areas, at the most basic level:
- The medical device
- Clinical evidence, suitably generated, appraised and analyzed
- Regulatory systems, processes and documents
Medical Devices
Medical devices must be appropriate for their intended use, have a positive benefit-risk ratio and meet all applicable MDR Annex I GSPRs. Devices must be produced under an appropriate quality management system (QMS). Most manufacturers use ISO 13485: 2016 as a standard to harmonize QMS suitability.
Clinical Evidence
All medical devices subject to the EU Medical Devices Directive (EU MDR) must be subject to the production and identification of clinical evidence. Conclusions on suitability of a device for its intended purpose (GSPR), GSPR compliance (benefit-risk profile) shall be supported by appropriately evaluated and analyzed clinical evidence. Clinical evidence includes evidence generated and stored by the manufacturer as well as independently published data in journals and elsewhere.
Regulatory Systems, Processes and Documents
The regulatory systems, processes and documentation required for MDR compliance in the EU are more comprehensive than those required under the out-of-print MDD. Requirements for MDR compliance include:
- QMS (Quality Management System) covering the entire organization
- PMS (Post-Market Surveillance) system monitoring the safety and performance of every device after it is placed on the market
- PMCF and vigilance system for every device
- Risk management system to the standard (ISO 14971:2019)
- Clinical evaluation process for each medical device documented in a well-structured CER (Clinical Evaluation Report) produced at regular intervals
- Dossier of technical documentation in accordance with the requirements of Annex II and Annex III
- A Person Responsible for Regulatory Compliance (PRRC) within an organization or permanently at his or her disposal.
Rules for evaluating compliance vary depending on the risk class of the device. For Class IIa, IIb, and III devices, notified bodies must perform an evaluation of the device, technical documentation, and regulatory systems to determine whether MDR compliance has been achieved. In contrast, most Class I devices typically do not require the involvement of a notified body, but must be supported by MDR-compliant regulatory files, systems, and processes. Class I products with sterile or metering capabilities must be approved by a notified body.
Compliance is evidenced by the CE marking on the device. Class I manufacturers (excluding products with sterile or measuring functionality) can apply the CE mark themselves after submitting a declaration of conformity. For all other classes of medical devices, the CE mark can only be affixed after official inspection and after the certification body has issued a certificate of conformity in accordance with the regulations of Chapter 4 of the MDR.
Effective way to achieve EU MDR Compliance
The most effective way to comply with the EU MDR consists of two different stages:
- Develop a deep understanding of MDR and related guidance documents.
- Apply this understanding to build a framework that meets your MDR requirements.
A deep understanding of the regulatory framework is an important first step to achieving MDR compliance. MDR describes a detailed regulatory environment that establishes rules and regulations for all aspects of medical device development, marketing, and performance monitoring. Therefore, it is important for medical device manufacturers to understand MDR in detail. The MDR introduces a number of changes that impose significant burdens on manufacturers in many areas.
Stricter Clinical Evidence Requirements
All available clinical evidence, whether favorable or unfavorable, must be identified in the context of the product in question and all comparable alternatives. Evidence must be evaluated and analyzed according to established protocols. In addition to traditional clinical evidence, PMCF systems must provide real-world evidence (RWE) that demonstrates the safe and effective performance of the device in the hands of “ordinary users.”
Equivalence Restrictions
Under the MDD, manufacturers can claim that their devices are “equivalent” to related devices already approved for sale and can then take advantage of this clinical evidence portfolio of comparable devices without having to create for own device. Under the MDR, the ability to do is severely limited.
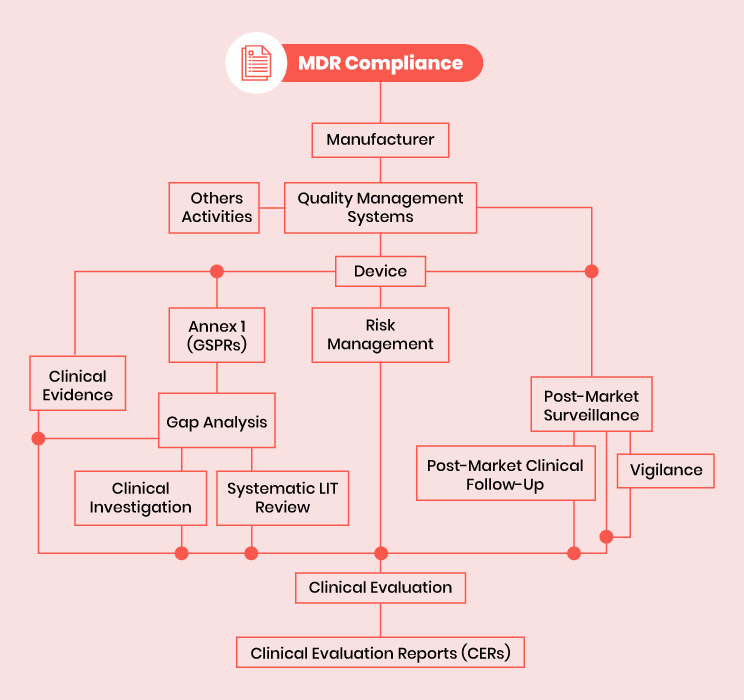
Fig 1: MDR compliance – The medical device regulation environment
It is also important to keep up to date with harmonized standards such as ISO standards. Compliance with ISO standards presupposes compliance with MDR in this area. Additionally, various MedDev guidelines provide guidance for structuring and implementing various MDR compliance activities.
Many of the regulatory processes required for MDR compliance follow the same circular structure for their implementation and maintenance, and this must be reflected in the design. This system typically includes:
- Establish
- Document
- Implement
- Update
- Improve
- Maintain
Applying this process to quality management systems, PMS systems, risk management processes, and clinical assessments ensures that the information collected during the Implementation phase is implemented. It was integrated into the design of the system and responded accordingly. Even with a good understanding of MDR, building a compliant system can be difficult. For example, demonstrating compliance with GSPR, developing PMCF procedures within PMS systems, and conducting clinical evaluations may include designing, documenting, implementing, conducting, and interpreting clinical investigations.
Implementation and interpretation of clinical evidence requires a level of medical acumen that many medical device companies do not have in-house.
How is the competition for MDR Compliance impacting the industry today?
Although the MDR is not yet fully in force, many manufacturers across all industries are facing the challenge of meeting EU MDR compliance requirements. To meet MDR requirements in a timely manner, many manufacturers must implement new guidelines and strategies long before the deadline. Long-held equivalence claims may no longer be valid, often meaning that a new evidence portfolio will need to be created from scratch. The need to substantiate every indication places significant demands on evidence generation systems, which must be expanded or redesigned to meet the requirements. It is estimated that almost 80% of European medical device companies do not yet have an adequate regulatory strategy to comply with the MDR. This highlights a lack of both industry understanding and willingness to comply with regulatory requirements. Many manufacturers must change their approach or risk having their products’ regulatory approvals revoked after the 2021 deadline.
Proposed Solution
Restrictions on Substances under REACH & CLP: The EU IVDR also restricts the use of substances covered by REACH (Registration, Evaluation, Authorization and Restriction of Chemicals) and CLP (Classification, Labelling and Packaging) Regulations, as is done in the case of the EU MDR. In order to comply with these regulations, manufacturers of IVD devices are required to identify and control substances of very high concern (SVHC) in their products. Manufacturers shall provide information to downstream users and consumers, including safe handling instructions and potential risks associated with the substance, if the SVHC exceeds 0.1% of the weight by weight.
In addition, harmonized criteria for classification, labelling and packaging of chemicals are laid down in the CLP Regulation. By correctly classifying dangerous substances present in their devices and providing appropriate labelling and safety information/data sheets to downstream users, medical device manufacturers shall comply with the CLP requirements.
Conclusion
For medical device manufacturers operating in the European Union, compliance with EU MDR and EU IVDR is of crucial importance. These regulations are intended to safeguard patients’ safety, harmonize standards and increase quality and performance of medical devices. The compliance requirements, including classification, technical documentation, involvement with notified bodies and restrictions on substances referred to in REACH and CLP shall be strictly complied with by manufacturers. This will allow manufacturers to navigate the regulatory environment, gain access to the market and contribute to the development of healthcare in the EU.
The EU MDR deadline has passed, and it is imperative that you ensure your medical devices adhere to the necessary requirements. Failure to comply may result in regulatory challenges and potential product recalls. Is your medical device EU MDR compliant? Evaluate its status promptly with the assistance of a local device regulatory expert. Contact MakroCare for expert consultation.
